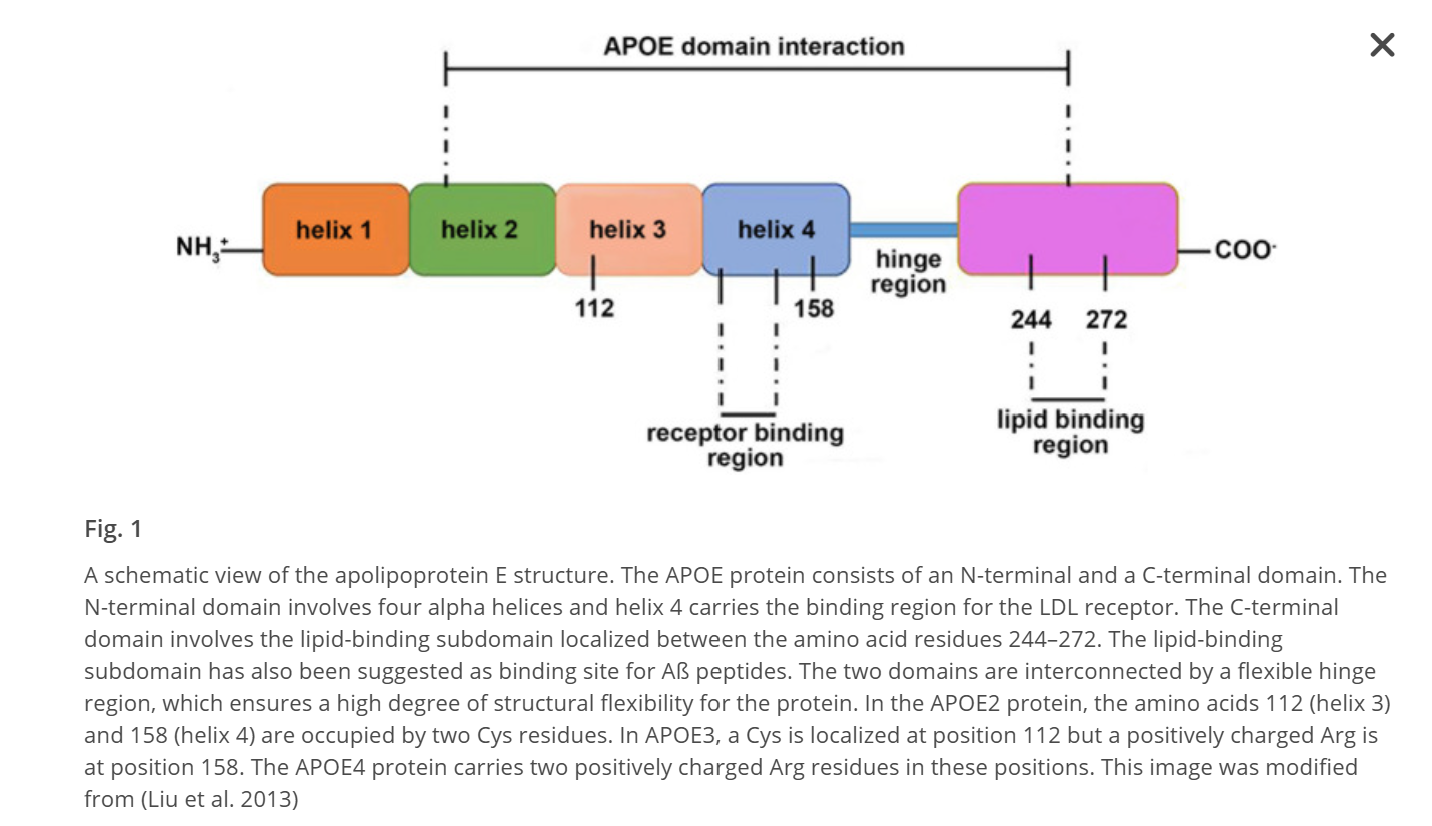
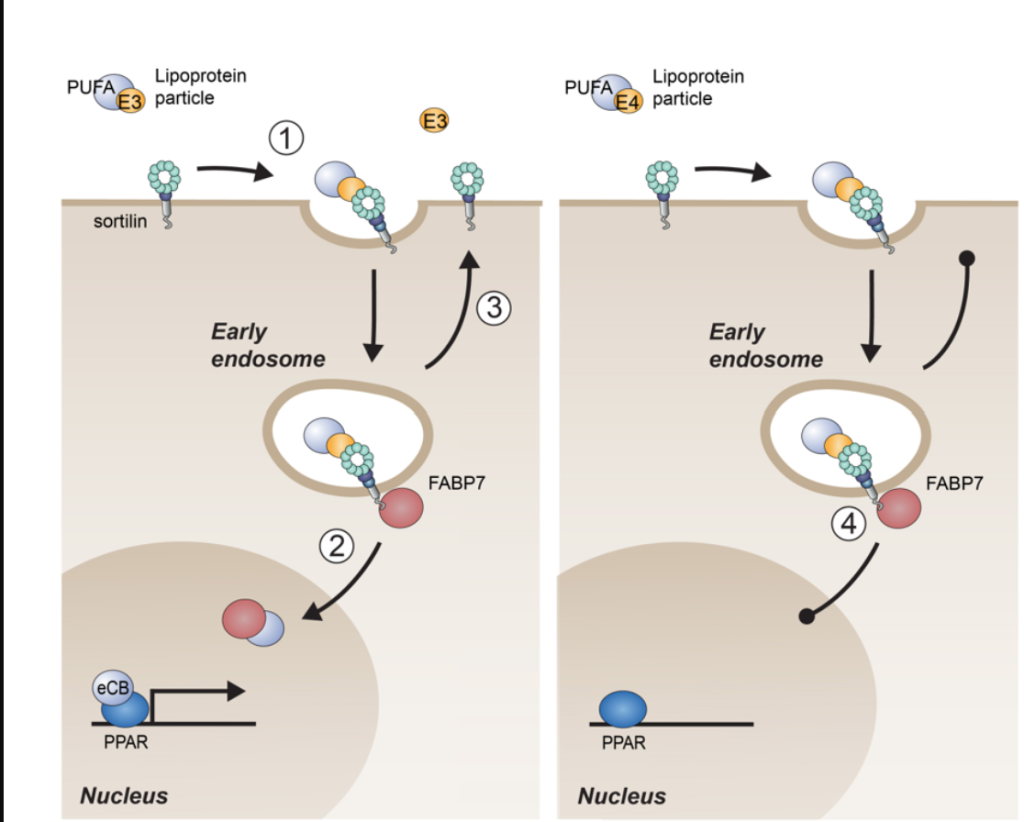
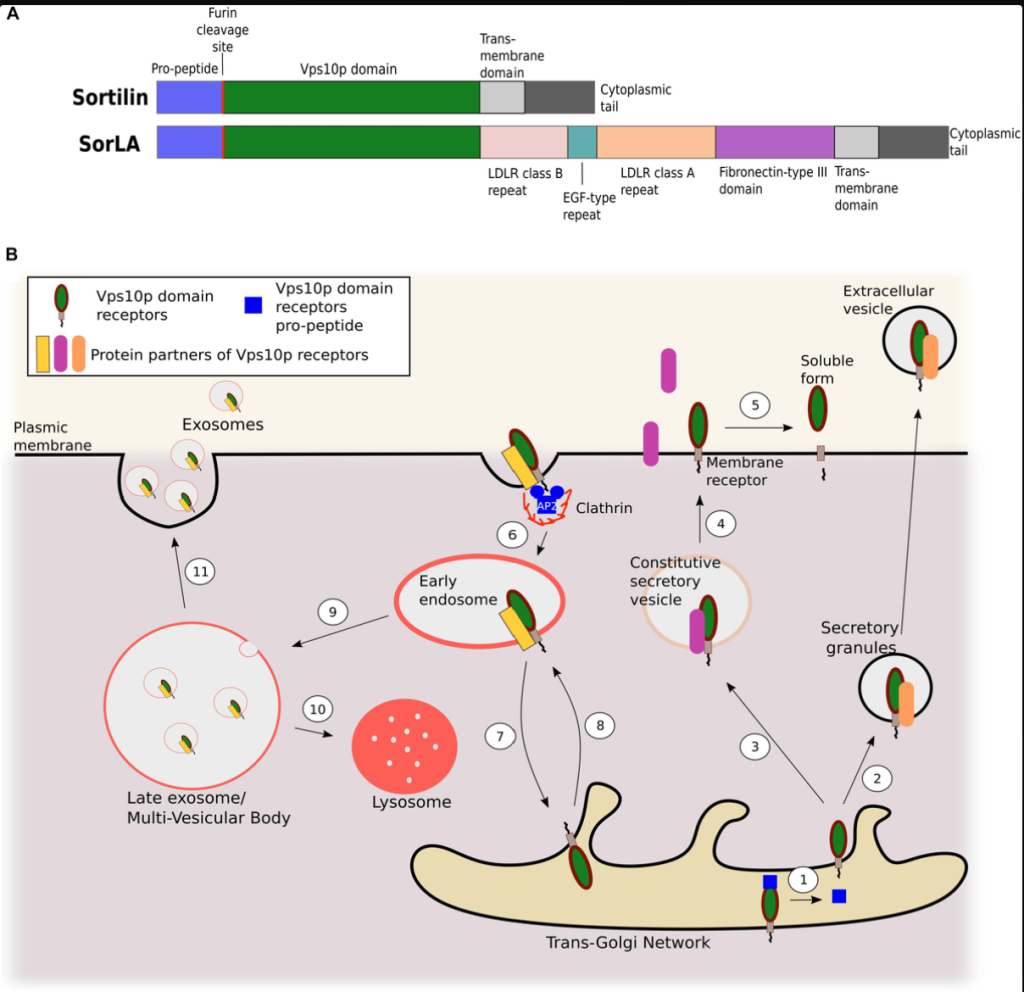
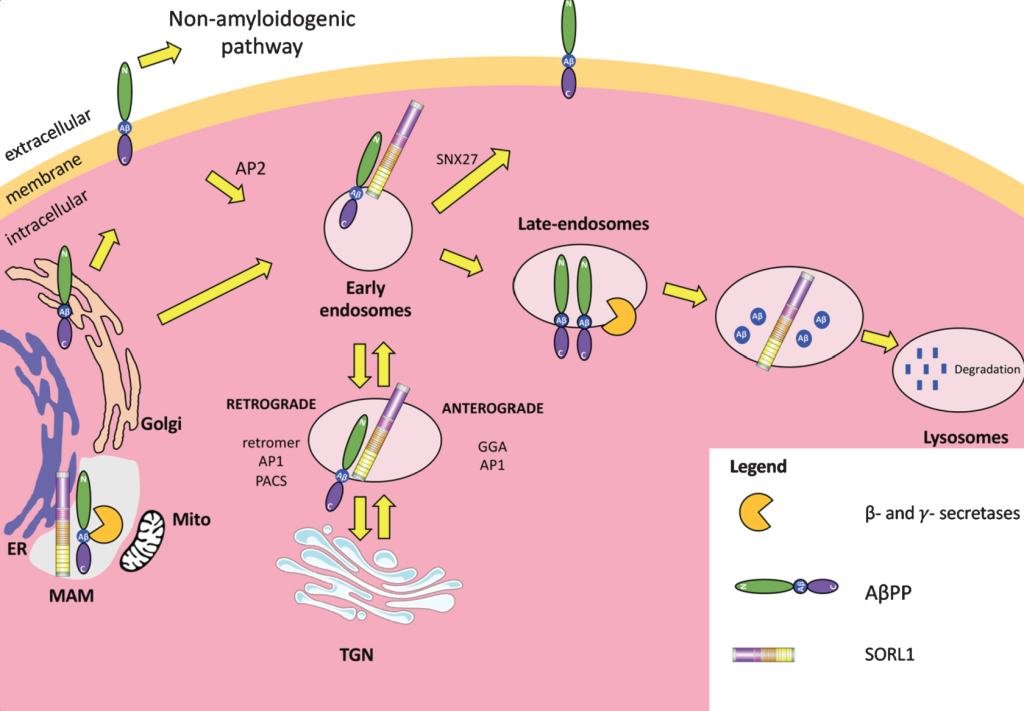
APOE4 reduces NPTX2 expression in neurons through several interconnected mechanisms that emerge early in Alzheimer’s pathogenesis. APOE4 carriers display a pronounced downregulation of NPTX2 at both mRNA and protein levels in the cortex, an effect that appears before overt neurodegeneration and is detectable in mild cognitive impairment. This reduction is not merely a consequence of neuron loss but reflects altered activity-dependent transcriptional regulation, impaired neurotrophic signaling, and increased repressive transcriptional pressure in vulnerable neurons.
Disruption of Activity-Regulated Gene Networks
NPTX2 is an immediate early gene crucial for synaptic function and the excitation-inhibition balance in neuronal circuits. Its expression is tightly coupled to neuronal activity and brain-derived neurotrophic factor (BDNF) signaling. In APOE4 and early Alzheimer’s, there is marked weakening of NPTX2’s correlation with BDNF, VGF, SST, and other genes that constitute the brain’s activity-regulated plasticity network. This points to a loss of the physiological mechanisms that normally drive NPTX2 transcription, likely resulting from both intrinsic neuronal dysfunction and loss of neurotrophic support.
Enhanced Inhibitory and Stress-Linked Regulation
Multi-omics analysis reveals that in AD—especially in APOE4 carriers—NPTX2 becomes increasingly negatively correlated with a cluster of transcriptional repressors and stress response genes (e.g., SMAD5, FOXJ1, ZIC4, ZHX3, JDP2). These factors are associated with chromatin remodeling and increased stress signaling. Their upregulation in AD produces a more repressive nuclear environment, further inhibiting NPTX2 transcription beyond what would be expected from synaptic or neuronal loss alone.
Dysfunctional Proteostasis and Mitochondrial Support
NPTX2 expression is also linked to mitochondrial metabolism and protein quality control (proteostasis). APOE4 influences both these processes—it is associated with mitochondrial dysfunction, and the breakdown of protein homeostasis appears to weaken NPTX2’s support network. These convergent failures undermine both the translation and stability of NPTX2 protein, compounding the deficits from disrupted transcription.
Summary Table
| Mechanism | Effect in APOE4/AD | Evidence |
|---|---|---|
| Loss of activity-dependent transcription | Decoupling of NPTX2 from BDNF/VGF/SST, reduced induction by neuronal activity | Transcriptomic/proteomic decorrelation, weakened synaptic module signatures |
| Increased transcriptional repression | Upregulation of stress-linked repressors, chromatin compaction | Protein-level negative correlation, enrichment of repressive pathways |
| Mitochondrial/proteostasis failure | Weakened association with mitochondrial, chaperone, and translation genes | Decreased mitochondrial pathway correlations, impaired proteostasis module |
Together, these changes explain why NPTX2 loss in APOE4-associated AD is an early, cell-intrinsic defect in neurons—not solely a marker of neurodegeneration but a nexus where neurotrophic support, transcriptional repression, and metabolic stress converge to diminish network resilience.