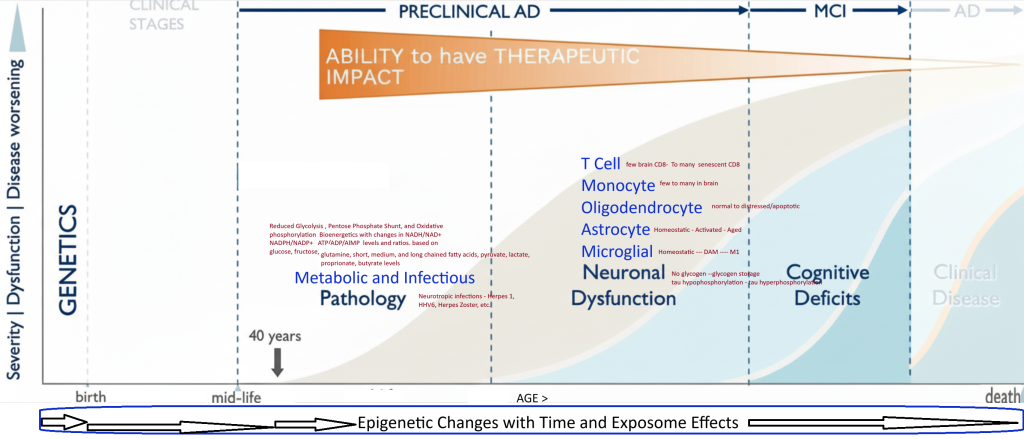
MCI and All Dementias : EOAD and LOAD
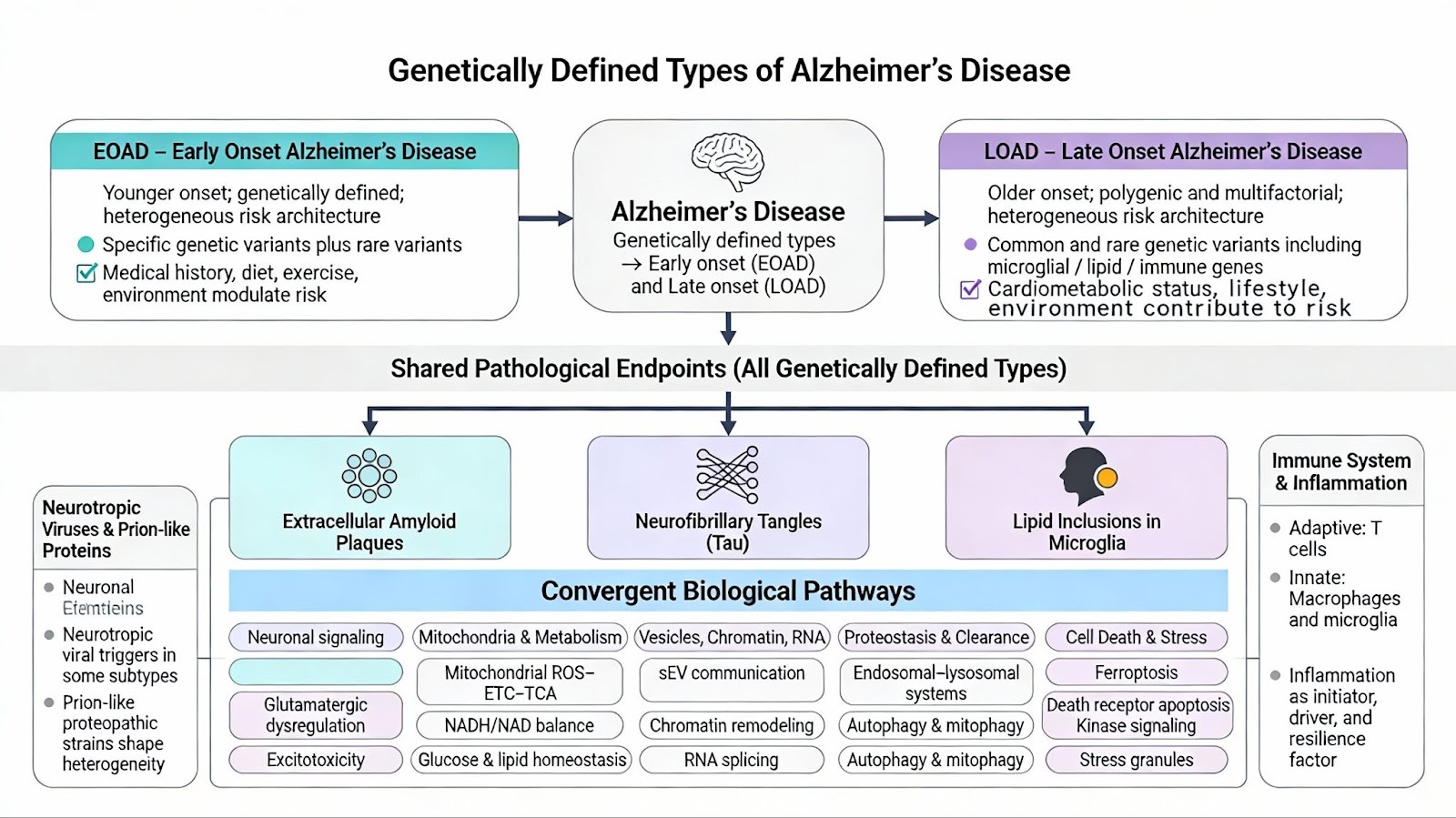
Genetically defined types of Alzheimer’s disease can be subtyped into either early or late onset. All of the reported genetically defined types share the common endpoint pathology of Neurofibrillary Tangles, Lipid inclusions in Microglia, and Extracellular Amyloid Plaques. Both LOAD and EOAD are heterogeneous diseases that are caused by specific genetic and exposome factors (, medical history, diet, exercise, and environmental factors). EOAD has about 90 % sporadic component and 10% familial, while LOAD has a 70% Polygenic component and a 30 % Sporadic subtype.
Framework 1- Complex Biological System Models with AD subtypes
Framework 2 –AD as an offshoot of the Biology of Aging
Framework 3 – The Multiple hit theory of Genetics, Oxidative stress, chronic inflammation, and disrupted electron energy homeostasis
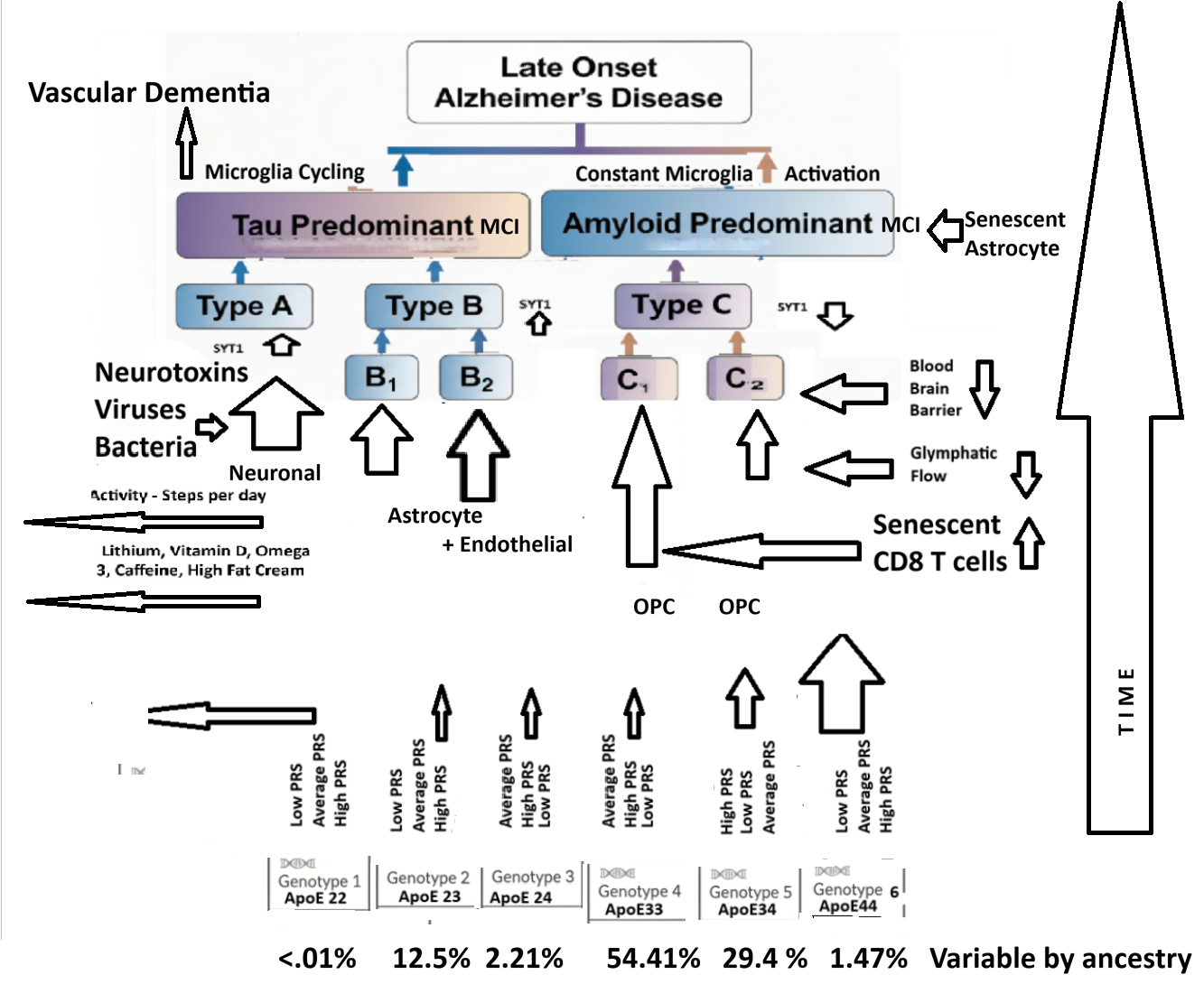
LOAD
Leading frameworks for the pathology of AD
Tau Predominant seen in microglial methylome data, brain transcriptome signatures, and Tau PET Scan analysis
33% A Neuronal dysfunction predominant – Most likely connected to Infection Related Sporadic AD of both Late and Early versions. May be related to Exitotoxicity – Mitochondrial dysfunction and Apoptosis pathway contributions.
33% B1 Astrocyte Predominant – Blood Brain Barrier and Glymphatic Flow – Amyloid Clearance connected and
B2 Endothelial then Astrocyte Predominant – CardioVascular Disease and Vascular Dementia connected
Amyloid Predominant seen in microglial methylome data and brain transcriptome signatures
33% C1 – OPC with High Microglial Contribution – Involving pathogenic CD8 T Cells and APoE4 influenced
C2 – OPC with Low Microglial Contribution – A primary Oliodendrocytopathy – not involving CD8 T cells.
ApoE genotype stratified patients differentially express proteomes leading to different LOAD Neuropathology Types depending on their Exposome and Polygenic Risk Factors. Exposome factors include HSV1, Lithium in diet, Mediterranean diet, Coffee consumption, High Fat Cream Consumption, Microbial Ferulic Acid Production in the Intestine, Physical Activity Level, Medications including levetiracetam- Keppra, Education Level, Social Experiences, Brain Exercises, Periodontitis, and/or other Risk Factors .
Pan ApoE genotype or ApoE independent Proteome Biomarkers
- Inflammation- ? IL-6 or interferon gamma from Local CD8 T cells or Senescent Astrocytes communicating with Microglia, and OPCs.
- Mitochondrial dysfunction- Due to ApoE4 and Hyperactivity of Excitotoxic Glutamate Neurons and Locus Ceruleus noradrenergic Neurons impacted by CD8 T cell binding to HLA 1
- Dyslipidemia- due to faulty transfer of FFA from ApoE4 to a FFA binding protein in late endosomes or mitochondria.
Th1 like app recognizing senescent CD8 T cells TREMs >>>> Traverse Blood Brain Barrier initially as CEMs – They then migrate into CSF accessible tissues to cause Neurodegeneration through binding to MHC Class 1 expressing Neurons located in Locus Ceruleus, Hippocampus and other Areas.
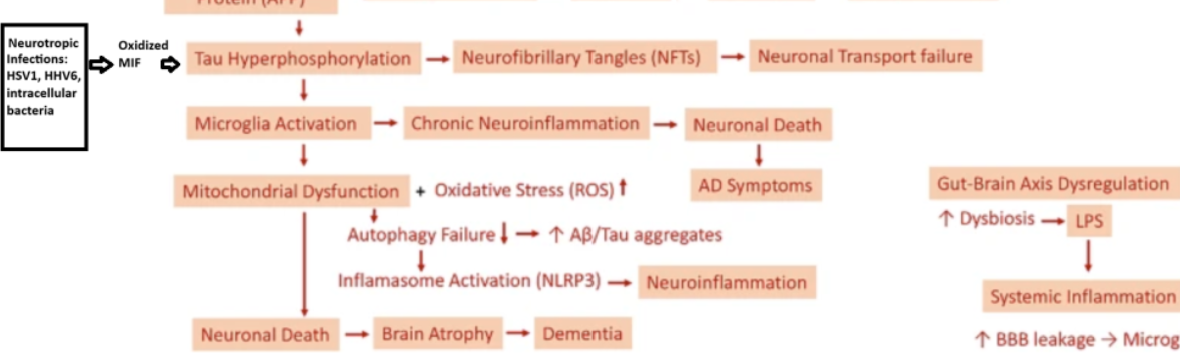
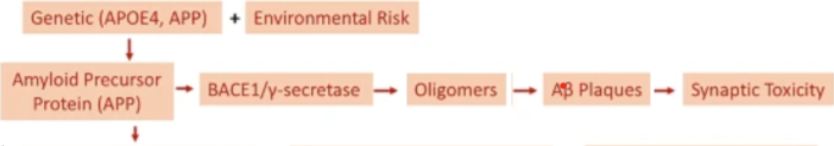
Pathological mechanisms and treatment progression of Alzheimer’s disease | European Journ
All Pathways